Нові підходи до визначення декстрану у процесі виробництва цукру

(За матеріалами журналу «Цукровий бізнес»», №2 (4), травень 2018 р.).
Наявність полісахаридів у сирому соку цукрового буряка негативно впливає на виробництво цукру, що, як правило, пом'якшується ферментативним розкладом декстранів. Цей вплив залежить не тільки від вмісту, але також і від молекулярно-масового розподілу. Тобто різні фракції декстрану по-різному впливають на виробничий процес. Отже, точний контроль процесу вимагає достовірних знань про наявний вміст та молекулярно-масовий розподіл декстрану. Детальне розуміння специфічних проблем переробки, а також цільового застосування ферментів вимагає визначення сумарного вмісту декстрану та його характеристик, включаючи диференціювання різних його фракцій. Для вирішення цієї проблеми були розроблені два нові підходи до визначення декстрану, які порівнювалися із загальноприйнятим газовим методом, що є досить неточним та чутливим до зміни молекулярної маси. Обидва підходи базуються на поляриметрії. Нові методи перевершують газовий метод, так як, враховуючи два варіанти молекулярної маси, вони дозволяють визначати ширший діапазон молекулярних розмірів, включаючи декстрани з низькою молекулярною масою.
Наявність полісахаридів у сирому соку цукрового буряка негативно впливає на виробництво цукру. Ці полісахариди можуть бути похідними рослинної біомаси або дії мікроорганізмів. Особливо це стосується декстрану, утвореного із залишків D-глюкози (декстрози) у результаті ферментативної активності кисломолочних бактерій, більш точно, зразками L. mesenteroides. Основний ланцюжок декстрану складається із залишків глюкози, зв'язаних з α-(1 →6). Точки розщеплення через α-(1 →3) глікозидних зв'язків можуть виникати до 5%, викликаючи відхилення від строго лінійної структури.
У виробництві цукрового буряка спостерігається наявність значного вмісту декстрану. У цьому випадку розвиток бактерій в основному ініціюється циклами заморожування-відтавання. В обох випадках декстрани екстрагуються разом із цукрозою та можуть по-різному впливати на виробництво цукру. Наприклад, наявність декстранів викликає підвищену в'язкість соків, що негативно впливає на швидкість фільтрації, випаровування та кристалізації.
У процесі очищення дифузійного соку модифікована форма та розмір осадженого кристалу карбонату кальцію, щепленого з нецукрами, як правило, погіршує процес фільтрації. Крім того, декстрани викликають модифікацію форми та розміру кристалів цукрози. Такі модифікації залежать не лише від існуючого вмісту, але також і від молекулярно-масового розподілу декстранів. Це означає, що вивчення впливу декстрану вимагає додаткового перегляду специфічних ефектів різних молекулярних масових фракцій. Отже, різні молекулярні маси тією чи іншою мірою можуть викликати безліч проблем із переробкою.
Відомо, що збільшення в'язкості, головним чином, обумовлено декстранами з високою молекулярною масою. Однак, специфіка вищезгаданих модифікаційних ефектів ще не деталізована. Добре відомо, що декстрани як із високою, так і з низькою молекулярною масою сприяють появі цих ефектів.
Отже, аналіз декстрану є необхідною передумовою для детального вивчення проблем із переробкою, які він викликає, а також для зменшення його впливу та модифікаційних ефектів контрольованим способом. Такі ефекти, як правило, пом'якшуються ферментативним розкладом декстрану, що призводить до поступового зменшення його молекулярної маси. Проте, деталі процесу розкладу та продуктів проміжної реакції, включаючи їх специфічні наслідки для цукрового виробництва, поки що необґрунтовано.
Правильний аналіз декстранів вимагає виконання двох основних вимог. Перша з них включає визначення загального вмісту декстрану, враховуючи декстрани з меншою молекулярною масою. Для цього потрібен сигнал відгуку, незалежний від молекулярної маси. Відомі способи в основному включають сигнал відгуку зі значною чутливістю до зміни молекулярної маси, що робить виявлення декстранів із високою та низькою молекулярною масою практично неможливим.
Для того щоб отримати найбільш точне уявлення про декстран, необхідно охарактеризувати розподіл молекул за їх величиною. Диференціація між декстранами з високою та низькою молекулярною масою є додатковою необхідною умовою для правильного проведення аналізу.
Проведення дослідів ускладнюється відносно низьким вмістом та широким діапазоном молекулярної маси дифузійного соку. Однак, встановлені методи визначення декстрану не відповідають жодній із цих вимог.
Використання спиртових осадів декстрану є популярним інструментом для аналізу полісахаридів. Загальноприйнятий газовий метод, згідно з ICUMSA, базується на спиртових осадах із використанням 50% (v/v) етанолу. Незважаючи на легкість виконання, сигнал відгуку цього методу та рівень спиртових осадів дуже чутливі до зміни молекулярної маси. Це призводить до досить неточного визначення вмісту декстрану, який додатково обмежується високою молекулярною масою.
Іншим затвердженим методом є мідний метод Робертса. У цьому випадку всі полісахариди осаджуються етанолом. Після цього декстран вибірково осаджується лужним сульфатом міді. Утворення кольорової суміші фенол-сірчаної кислоти визначається за допомогою спектрофотометру (485 нм). Проте, нестача часу та хімічних речовин роблять цей метод непридатним для промислового використання. Крім того, немає точної межі між розміром молекул та кількістю осадженого декстрану з використанням навіть 80% етанолу. Раніше отримані результати на основі поляриметрії показали, що з використанням 80% етанолу низькомолекулярний декстран із середньою молекулярною масою 40 кДа не повністю відновлюється.
Це питання також стосується інших методів використання спиртових осадів у якості підготовчого етапу, наприклад, метод ферменту-HPLC (високоефективної рідинної хроматографії (ВЕРХ)), в якому використовуються спиртові осади та 80% етанолу для ізоляції усіх полісахаридів. Під дією ферментів декстран розкладається до продуктів кінцевого розпаду — ізомальтози (декстринози). Після цього вміст ізомальтози використовується для розрахунку вихідного вмісту декстрану. Так чи інакше, розпад виключно на ізомальтозу є сумнівним. Поява розгалуження у ланцюзі є найбільш очевидним фактором, який сприяє розпаду суміші олігосахаридів та ізомальтози. Основним недоліком цього методу є його невідповідність промисловій практиці, оскільки у процесі його виконання застосовується хроматографічне обладнання та витрачається багато часу.
Отже, досі немає ідеального індустріального методу для визначення загального вмісту декстранів із різним діапазоном молекулярних розмірів. У процесі пошуку нових методів було проведено порівняльні дослідження та проаналізовано газовий метод.
Нові підходи базуються на поляриметрії, що вимагає використання стандартного промислового обладнання. Передбачається, що сигнал відгуку оптичного обертання є незалежним від молекулярної маси декстранів. Вивчення аналітичних методів визначення вмісту декстранів вимагає детального розуміння сигналу відгуку та потенційних змін у параметрах процесу переробки або інших компонентів. Ступінь та сила сигналу є вирішальними для визначення значущих діапазонів щодо вмісту та розподілу молекулярних мас декстранів.
Матеріал та метод
У роботі використовувались різні молекулярно-масові фракції декстрану. Середню молекулярну масу декстрану (Carl Roth, декстран 500) використовували для підготовки калібрувального графіку всіх застосованих методів. Високо- (Sigmar-Aldrich, декстран 2000 з розподілом молекулярної маси від 150000 до 2800000 D2) та низькомолекулярні декстрани (Carl Roth, декстран 40 з розподілом молекулярної маси 35000-45000 D3) були додатково використані для одержання штучних сатураційних соків, що містять цукрозу 15 г/100 г. Усі фракції декстрану походять від кисломолочних бактерій, а саме від штаму бактерій Leuconostoc mesenteroides B512. Технічні характеристики виробників свідчать про дуже подібні обертання площини поляризації різних фракцій декстрану. Наприклад, фракції з високою молекулярною масою мають обертання площини поляризації при температурі +199°. Середні та низькомолекулярні декстрани мають цілком подібні значення для обертання площини поляризації з +195° до +203° та з +195° до +201° відповідно.
Ви значення декстрану
|
|
|
|
|
 Газовий метод
Газовий метод Ферментативний метод
Ферментативний метод Мембранний метод
Мембранний метод Поляриметричні вимірювання
Поляриметричні вимірювання Хроматографічні вимірювання
Хроматографічні вимірюванняОбговорення результатів
Газовий метод
Газовий метод у даний час є найпоширенішим методом для аналізу декстрану в процесі виробництва цукру. Він базується на спиртових осадах та фотометрично вимірює отриману мутність. Водорозчинні полісахариди, такі як декстран, взаємодіють з молекулами води через водневі зв'язки. Відповідно, наявний етанол порушує взаємодію декстрану з водою, внаслідок чого вдається визначити мутність. Розчинність полісахаридів залежить переважно від їх молекулярної структури та маси, що виражається у зменшенні насиченості та збільшенні молекулярної маси. Отже, ступінь спиртових осадів не узгоджується з коливаннями молекулярної маси.
Чутливість сигналу відгуку до різних фракцій декстрану викликає неточне визначення декстрану. Отже, цей метод може бути корисним для специфічних наборів зразків з обмеженими варіаціями розмірів молекул. У цьому випадку необхідним є використання відповідного матеріалу для калібрувального графіку. Однак, це неможливо, якщо розподіл за величиною молекул зразків, що аналізуються, не визначається апріорно. Таким чином, вибір калібрувального матеріалу є вирішальним для визначення декстрану. Тому у цей спосіб неможливо вирішити проблему підготовки калібрувального графіка.
Кількість етанолу, що використовується для отримання мутності, впливає на насиченість а, отже, й на процес осадження. Відповідно до Міжнародного комітету по загальноприйнятим методам аналізу цукру (ICUMSA), однакова аліквота етанолу використовується для формування імпульсу (50% (v/v) етанолу). Основна мета — уникнути осідання інших нецукрів нижчої молекулярної маси, таких як пектини. Однак, це є основною причиною їх обмеження в декстранах з високою молекулярною масою. Крім того, газовий метод не гарантує отримання інформації про молекулярний склад усіх декстранів. Незважаючи на це, раніше отримані результати показали, що комбінації методів, таких як газового та мідного методу Робертса, можуть бути корисними для орієнтовного визначення характеристик молекулярно-масового розподілу. Однак, ця комбінація не підходить для промислового застосування, оскільки мідний метод Робертса є обтяжливим та хімічно трудомістким.
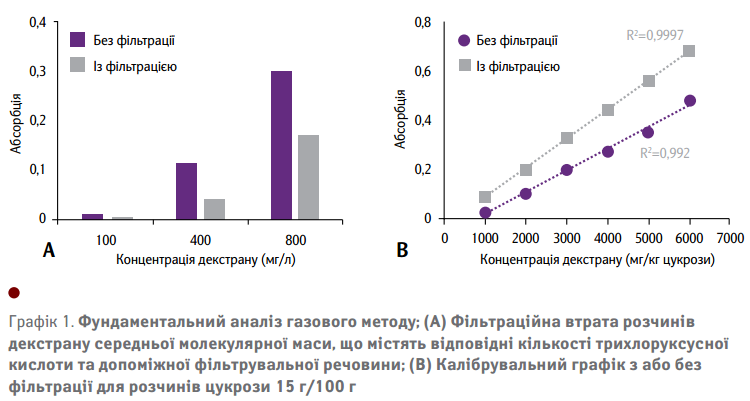
Газовий метод включає вилучення крохмалю та білків з подальшою фільтрацією до фактичного спиртового осаду. Відповідно до методу ICUMSA, при визначенні калібрувальних графіків етап фільтрації упускається. Вплив фільтрації на результати газового методу показано на Графіку 1(А). Розчини, що містять відповідну кількість декстрану, трихлоруксусної кислоти та допоміжної фільтрувальної речовини, були проаналізовані до та після фільтрації.
Зміни у процесі абсорбції показують значну втрату декстрану під час фільтрації. Це пов'язано з адгезією внутрішнього отвору у фільтрі та частинок фільтрувального порошку. На Графіку 1(B) показано відмінності між встановленням калібрувальної кривої з або без фільтрації, що підходить для аналізу розчинів цукрози 15 г/100 г. Дані свідчать, що пропускання етапу фільтрації під час калібрування може призвести до суттєвого заниження оцінки вмісту декстрану. Таким чином, доцільно запровадити відповідні зміни у процесі калібрування. Сила адгезії між молекулами, які належать до різних типів, зокрема, пов'язана з розмірами та геометрією контактних ділянок. Проте, газовий метод є широко використовуваним методом визначення декстрану у процесі виробництва цукру. Тому, саме він став еталоном для розробки двох нових підходів до визначення декстрану.
Ферментативний метод
Ферментативний метод засновано на зміні оптичного обертання внаслідок повного ферментативного розпаду декстрану. Загалом, вважається, що декстрани з високою та низькою молекулярною масою мають однакове оптичне обертання, що робить сигнал відгуку незалежним від молекулярно-масового розподілу. Передбачається, що продукти розпаду складаються з оліго-сахаридів та ізомальтози залежно від способу дії ферменту та ступеня розгалуження субстрату та мають нижче оптичне обертання, порівняно з декстраном. Різниця в оптичному обертанні між початковим розчином декстранів та розчином, що містить продукти розпаду, стосується вихідного вмісту декстрану. Щодо методологічних експериментів в оптимальних умовах, ферментативна реакція постійно контролювалася у різних умовах. Із цією метою були проведені експерименти з гідролізу чистих розчинів декстрану у поляриметровій трубці для того, щоб визначити зміни оптичного обертання під час ферментативного розпаду з плином часу.
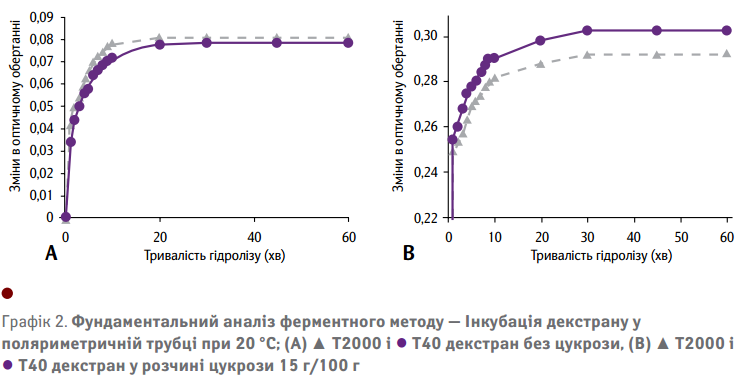
Температура у трубці становила 20 °С відповідно до обмеження температури поляриметру. Як показано на Графіку 2(А), оптичні обертання під час ферментативного гідролізу водних розчинів декстрану з високою та низькою молекулярною масою практично ідентичні. Це підтверджує основне припущення відносно незалежності сигналу відгуку щодо молекулярної маси. Отже, це означає, що сигнал відгуку практично не залежить від варіації молекулярної маси вихідного матеріалу у чистих розчинах декстрану. Це є основною перевагою перед газовим методом, що потенційно дає змогу визначити загальний вміст декстрану. Крім того, на Графіку 2(А) показано, що після 20 хв, коли використовується достатній надлишок ферменту при температурі інкубації 20 °С, ніяких змін в оптичному обертанні не відбувається.
Раніше проведені досліди показали, що наявність цукрози впливає на ферментативну реакцію, що свідчить про можливе неконкурентне інгібування.
Реакція ферменту-каталізатора зазвичай поділяється на три основні етапи, що включають утворення фермент-субстратного комплексу, фактичну каталізовану реакцію та остаточний розпад продукту. Специфічне зв'язування субстрату з активним ферментом є необхідною умовою реакції ферментів-каталізаторів, яка ґрунтується або на принципі ключів, або на моделі індукованої відповідності. В обох випадках кінцевий вигляд активного ферменту доповнює позицію установки субстрату.
Конформаційні зміни субстрату внаслідок присутності інших складових у розчині можуть викликати зміни фермент-субстрат-зв’язків. Потенційно це призводить до зміни тривалості ферментативної реакції або типу та кількості продуктів розпаду, що негативно впливає на загальну зміну оптичного обертання внаслідок ферментативного гідролізу.
Для того щоб визначити, яким чином наявність цукрози підтверджує ферментативний розпад декстрану, додатково проводилися експерименти з гідролізу, що містили цукрозу 15 г/100 г. Як видно на Графіку 2(В), ці експерименти показали подібні результати до тих, що мають чисті розчини декстрану (Графік 2(А)). Дані на Графіку 2(В) показують, що повне розчеплення декстранів з високою та низькою молекулярною масою у розчині цукрози 15 г/100 г вимагає трохи більше часу (30 хв). У цьому випадку в оптичному обертанні відбуваються відносно значні зміни.
Це викликано частими побічними ефектами під час розпаду з використанням надзвичайно високого вмісту ферментів. Щоб зменшити ефекти цієї побічної активності на фактичне вимірювання, проводилася повторна перевірка. Це стосується технологічного процесу, а також складу зразків. Отже, фіксується постійний вміст цукрози 15 г/100 г для процесу перевірки та вимірювання, що одночасно викликає зміни в оптичному обертанні.
На відміну від чистих розчинів декстрану, зміна оптичного обертання декстанів із низькою та високою молекулярною масою через ферментативне розщеплення, здається, дещо відрізняється у контексті наявності цукрози. Молекулярна маса субстрату впливає на схему реакції ферменту. Потрібно детально описати потенційні конформаційні зміни молекул декстрану у розчині. Щоб звести до мінімуму можливі ефекти розміру молекул декстрану, калібрувальна крива знову враховувала середню молекулярну масу декстрану точно так само, як для інших методів. Проте, основний аналіз проводився з використанням оптимальних умов ферментативної реакції для забезпечення повного ферментативного розпаду. Таким чином, було встановлено оптимальну температуру для дії декстранази 55 °С та час інкубації 60 хв. Отже, при цій температурі ферментативна реакція відбувається швидше. На цьому етапі слід зазначити, що для зменшення часу підготовки зразків потрібно змінити параметри ферментативної реакції. Також слід підкреслити, що зміна оптичного обертання внаслідок ферментативного розчеплення є порівняно невеликою, що вимагає обережного поводження та використання дуже точних кругових поляриметрів.
Мембранний метод
Мембранний метод має за основу добре встановлений метод утримання макромолекул механічними засобами. Вплив фільтрації на фільтрат по відношенню до вихідного розчину знову визначається за допомогою поляриметру. Принцип цього методу дуже подібний до ферментативного методу. Основна відмінність полягає у тому, що замість ферментативного розчеплення відбувається поділ декстрану через мембранну фільтрацію, у результаті чого визначається зміна в оптичному обертанні початкового розчину та фільтрату. Ця різниця потім використовується для розрахунку вмісту декстрану у початковому розчині. Сигнал відгуку поляриметру не залежить від молекулярно-масового розподілу декстрану.
Процес поділу через мембранну фільтрацію залежить від характеристик мембрани та розчину. Ключовим чинником поділу є взаємозв'язок між розміром пор мембран, розподілом форми та структури полімерів, точніше виглядом молекул та гідродинамічним радіусом у розчині компоненту, що відокремлюється.
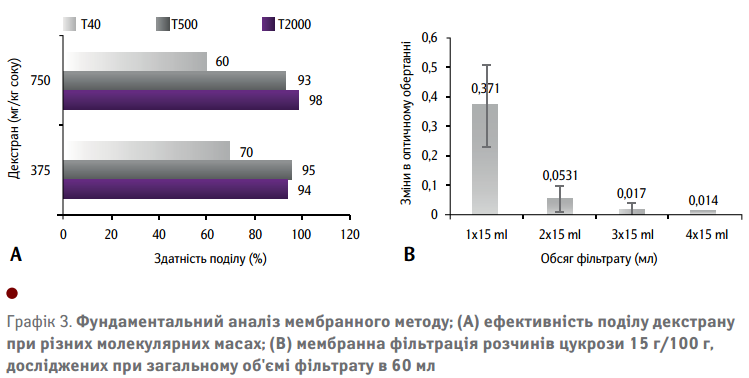
Графік 3(А) показує вплив варіації розмірів молекул на процес поділу у чистих розчинах декстрану. Результати свідчать про майже 100%-у ефективність поділу для високо- та середньомолекулярних фракцій у чистих розчинах декстрану. На відміну від цього, низькомолекулярний декстран показує ефективність поділу приблизно 60-70% у чистих розчинах. Глікозидні зв'язки, однак, розширюються і дають змогу утворювати різні конформаційні структури, що передбачають гнучку просторову структуру полімерного ланцюга, зокрема, глікозидові зв'язки у первинній гідроксильній групі, як правило, більш розповсюджені, що вказує на вищу щільність α-1,6-зв’язки у молекулах декстрану. Отже, припускається, що вища молекулярна маса декстану має характеристики розширюваної статистичної спіралі.
Залежно від молекулярної маси існує конформаційний перехід стану полімеру, що призводить до невпорядкованої конформації високомолекулярних декстранів та витягнутої стержнеподібної конформації молекул із низькою молекулярною масою. Ця стержнеподібна конформація низькомолекулярного декстрану та, зокрема, нижня межа цієї полідисперсної фракції описують здатність лінійних молекул проходити пори мембрани, незважаючи на те, що характеристика мембран та молекулярна маса вказують протилежне.
Добре відомо, що як і мембрана, так і конформація полімеру можуть змінитися у зв’язку з наявністю інших компонентів у розчині. Знову ж таки, прищеплення основних компонентів розчину до внутрішньої поверхні пор мембран, ймовірно, відбувається аналогічно ефектам адгезії під час етапу фільтрації за газовим методом.
Більш гладкі внутрішні стінки мембран мають більшу чутливість до зчеплення. Навіть незначне збереження оптично активної цукрози у мембрані сприяє різниці оптичного обертання між початковим розчином та фільтратом.
Вплив наявної цукрози оцінювався у процесі аналізу змін в оптичному обертанні між початковим розчином і фільтратом чистих розчинів цукрози при загальному вмісту фільтрату 60 мл. Було відібрано аліквоти фільтрату у межах 15 мл, які використовувалися для визначення різниці оптичного обертання, порівняно з розчином цукрози 15 г/100 г початкового зразка. Існує істотна різниця в оптичному обертанні початкового розчину та фільтрату, що чітко вказує на зчеплення цукрози з мембраною. Ця різниця згодом зменшується у процесі фільтрації. Щоб зменшити вплив цукрози, перша аліквота фільтрату була відкинута. Отже, для фактичного вимірювання використовувалася друга аліквота. Було визначено середню різницю в оптичному обертанні 0,05° для другої аліквоти фільтрату чистих розчинів цукрози 15 г/100 г (див. Графік 3(B)). Це значення відповідає приблизно 700 мг декстрану на 1 кг цукрози. Таким чином, використовуючи цю аліквоту, все ще видно невелику різницю в оптичному обертанні, викликану наявною цукрозою. Додатковим засобом для усунення впливу цукрози є включення стадії мембранної фільтрації у процес калібрування, що також враховує потенційну залежність цукрози від наявного вмісту декстрану. Зразки знову стандартизували до постійного вмісту цукрози 15 г/100 г. Крім того, для підготовки калібрувальної кривої була використана середня молекулярна маса декстрану.
Визначення вмісту декстрану та молекулярної маси
Визначивши основи кожного аналітичного методу, наступні дані щодо визначення декстрану повинні підтвердити свою придатність до аналізу різних молекулярних мас декстранів у розчинах цукрози. На Графіку 4(А) показано результати дослідження зразків, що містять високомолекулярний декстран у синтетичних сатураційних соках із використанням трьох різних способів, описаних вище. Всі методи використовують калібрувальні криві, які враховують середню молекулярну масу декстрану.
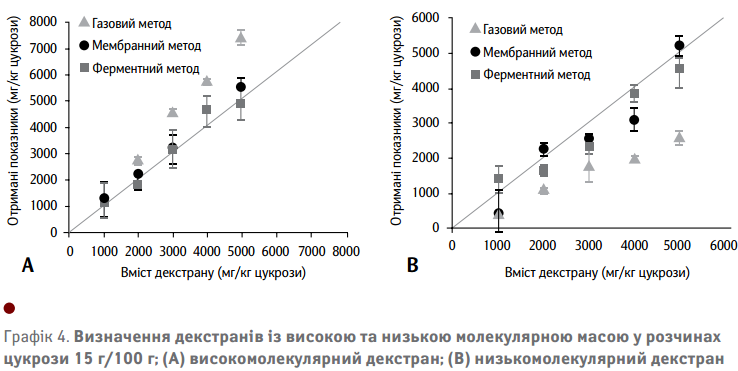
Газовий метод значно переоцінює високомолекулярний декстран. Це підтверджує вищезгадану чутливість цього методу до варіацій молекулярних мас через різницю в їх розчинності. Визначення вмісту декстрану з низькою молекулярною масою показано на Графіку 4(В). Газовий метод, очевидно, недооцінює низькомолекулярний декстран, що повністю узгоджується з попередніми міркуваннями. Цей широко використовуваний аналітичний метод має досить обмежену здатність визначення загального вмісту декстрану з урахуванням коливань молекулярної маси.
Визначення високомолекулярного декстрану з використанням ферментативного та мембранного методів показує вміст декстрану дуже близький до фактичних значень підготовлених розчинів (Графік 4 (A)). Визначення вмісту декстрану з низькою молекулярною масою (Графік 4 (B)) також краще вимірюється, порівняно з газовим методом. Обидва методи дають значення, близькі до фактичного вмісту, але дані свідчать про меншу точність, ніж для високомолекулярних декстранів.
Хороші результати, отримані ферментативним методом, є дещо незрозумілими, оскільки, як виявляється, це обумовлено не лише впливом конформації ферменту та декстрану, але також і дією на нижньому кінці поляриметричного діапазону вимірювань. Більш точне вимірювання оптичного обертання може зменшити відхилення та покращити отримані результати.
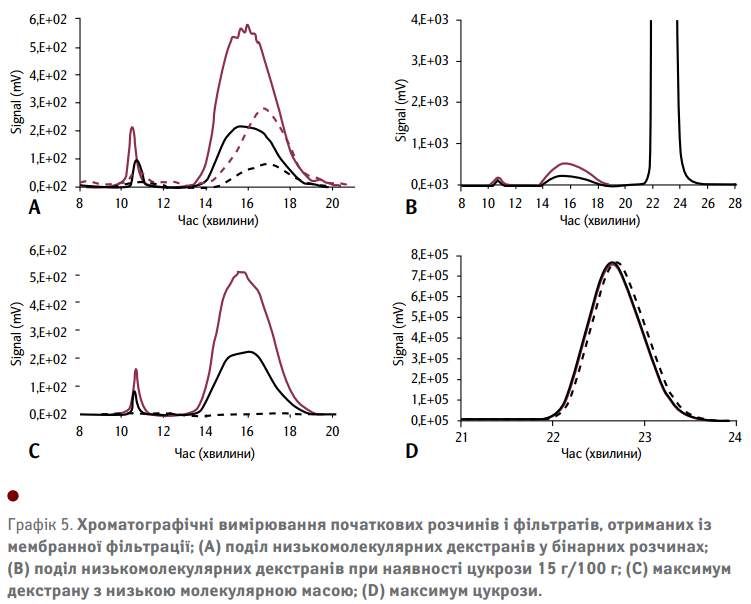
Чорні крапки на Графіку 4(В) являють собою визначення вмісту декстрану з низькою молекулярною масою у синтетичних сатураційних соках за допомогою мембранного методу. Отримані дані показують вищий вміст декстрану, ніж можна було очікувати від аналізу ефективності поділу в бінарних розчинах декстрану (див. Графік 3 (А)). Це може бути викликано вищим вмістом або цукрози, або декстрану. Для кращого розуміння таких ефектів, проводилися хроматографічні вимірювання початкових розчинів та фільтрату мембранно-фільтрованих розчинів декстрану з та без цукрози. На Графіку 5(А) показані хроматограми для початкових розчинів та фільтратів бінарних розчинів із різним вмістом декстрану. Отримані дані підтверджують вищезгадане неповне відокремлення низькомолекулярного декстрану в бінарних розчинах. Це пояснюється наявністю великої кількості декстрану у хроматограмах фільтрату, зображеного пунктирною лінією.
Інші компоненти розчину можуть змінювати структуру пор мембран, а також конформацію полімерів через специфічні взаємодії. Хроматографічний аналіз синтетичних сатураційних соків, що містять 15 г/100 г цукрози, показано на Графіку 5(В). Зокрема, максимум декстрану зображено на Графіку 5(С), що вказує на повне утримання низькомолекулярних декстранів, присутніх у початковому розчині, пунктирна крива збігається з базовою лінією та підтверджує кращу ефективність поділу фракцій декстранів з низькою молекулярною масою через наявність цукрози.
Це, швидше за все, викликано конформаційною різницею між стержневидною структурою декстрану у водному розчині та більш громіздкою сферичною структурою у розчині цукрози через специфічні взаємодії цукрози з декстраном. Наявність інших вуглеводів впливає на можливі «кругові» ефекти, так як вони можуть перешкоджати або повністю придушувати ці ефекти через низькомолекулярні декстрани.
Червона суцільна лінія (початковий розчин) та червона пунктирна лінія (розчин фільтрату), що містить 5000 мг декстрану на кг цукрози; чорна суцільна лінія (початковий розчин) і чорна пунктирна лінія (розчин фільтрату), що містить 3000 мг декстрану на кг цукрози.
Як зазначалося вище, потрібно приділяти більше уваги можливим змінам характеристик мембран. Це стосується осідання молекул цукрози на поверхню мембрани та/або на внутрішні стінки пор мембран. Можливе виникнення звужених або навіть заблокованих пор впливає на проникність мембрани. Якщо первинна структура пори гетерогенної мембрани не пошкоджена, декстран або, можливо, цукроза можуть накопичитися на поверхні мембрани, що часто впливає на сам процес фільтрації. З огляду на збільшення кількості цукрози (Графік 5(D)), неможливо виявити помітну зміну вмісту цукрози між початковим розчином та фільтратом. Це підтверджує, що ефекту накопичення цукрози можна запобігти, відкидаючи першу аліквоту фільтрату. Передбачалося, що зміни конформацій полімеру, індуковані цукрозою, відіграють певну роль як у модифікації кінетики ферментативних реакцій для декстранів з високою і низькою молекулярною масою, так і утримування мембран низькомолекулярних декстранів. З одного боку, важливо перевірити або, точніше, зрозуміти припущення про цю штучну конформаційну зміну для розробки надійних аналітичних методів. З іншого боку, більш детальні знання про це явище дозволять обміркувати кільцевий розвиток, щоб краще зрозуміти процес мембранного поділу у цілому. Проте, ферментативний та мембранний методи виявилися ефективнішими за газовий метод у процесі визначення вмісту декстрану з урахуванням різних молекулярних масових фракцій. Достовірне визначення широкого діапазону молекулярних розмірів є необхідною умовою будь-якого розширеного аналізу декстрану.
ВисновкиДва нові підходи до визначення вмісту декстрану — ферментативний і мембранний методи — було розроблено та порівняно з широко використовуваним газовим методом. Обидва методи засновані на визначенні відмінностей в оптичному обертанні і, отже, вимагають використання високочутливих кругових поляриметрів. Доведено, що поляриметрія не залежить від розподілу за величиною молекул декстрану. Звідси випливає, що ці методи є ефективними для кількісного визначення повного розмірного ряду молекул декстрану. Прототипи нових методів показують, що вони придатні до застосування у промисловій практиці, так як тип обладнання, тривалість проведення та споживання хімічних речовин знаходяться в розумних межах. Детальна оцінка цих методів показала, що включення всіх етапів підготовки зразків у процес встановлення калібрувальних кривих, як правило, є корисним для уникнення дефектів. Крім того, робота з фіксованим вмістом цукрози у процесі калібрування та аналізу підвищує точність даних підходів, тому що методи на основі поляриметрії чутливі до коливань вмісту цукрози. Крім того, припускається, що наявність цукрози викликає конформаційні зміни розчинних декстранів. Виявляється, що продукти, отримані у результаті повного ферментативно-каталізованого гідролізу декстрану, мають різний вміст цукрози у високо- та низькомолекулярних декстранах. У водних розчинах декстрану цієї різниці не було. Наявність цукрози також викликала утримання менших лінійних декстанів мембранами, що значно покращило ефективність поділу. Збільшення ефективності поділу від 60% до практично 100% було підтверджено хроматографічними вимірами. Зібрані дані підтверджують, що нові методи визначення вмісту декстрану перевершують газовий метод, особливо при тому, що декстран має значні коливання молекулярної маси. Це успішний крок до надійного та практичного методу, який дає змогу більш ефективно обмежити та попередити забруднення декстраном. |